-
Miyamoto D, Santi CG, Aoki V, Maruta CW. Bullous pemphigoid. An Bras Dermatol. 2019;94(2):133-146.
DOI: no disponible en resumen. PMID: 31090818.
Enlace PubMed
Resumen breve: Revisión completa y muy práctica sobre epidemiología, clínica, patogenia, diagnóstico y tratamiento del PA, con énfasis en las formas no ampollosas y en la realidad de países en desarrollo.
-
Bernard P, Antonicelli F. Bullous pemphigoid: A review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18(4):513-528.
DOI: 10.1007/s40257-017-0271-7. PMID: 28247089.
Enlace PubMed
Resumen breve: Revisión de referencia que resume criterios diagnósticos, asociaciones (neurológicas, fármacos) y estrategias terapéuticas, con enfoque práctico en pacientes ancianos.
-
Kridin K, Ludwig RJ. The growing incidence of bullous pemphigoid: Overview and potential explanations. Front Med (Lausanne). 2018;5:220.
DOI: 10.3389/fmed.2018.00220. PMID: 30177969.
Enlace PubMed
Resumen breve: Revisión epidemiológica que documenta el incremento de incidencia de PA y discute causas potenciales (envejecimiento, fármacos, mejora diagnóstica).
-
Fuertes de Vega I, Iranzo-Fernández P, Mascaró-Galy JM. Bullous pemphigoid: clinical practice guidelines. Actas Dermosifiliogr. 2014;105(4):328-346.
DOI: no disponible en resumen. PMID: 23540594.
Enlace PubMed
Resumen breve: Guía práctica en español sobre diagnóstico y tratamiento del PA, con algoritmos claros e indicaciones sobre biopsia, DIF, manejo tópico y sistémico.
-
Genovese G, Di Zenzo G, Cozzani E, et al. New Insights Into the Pathogenesis of Bullous Pemphigoid: 2019 Update. Front Immunol. 2019;10:1506.
DOI: 10.3389/fimmu.2019.01506. PMID: 31312206.
Enlace PubMed
Resumen breve: Actualización profunda sobre mecanismos inmunológicos del PA (complemento, IgE, eosinófilos, mastocitos, citocinas) y su posible repercusión terapéutica.
-
Cole C, Vinay K, Borradori L, Amber KT. Insights Into the Pathogenesis of Bullous Pemphigoid: The Role of Complement-Independent Mechanisms. Front Immunol. 2022;13:912876.
DOI: 10.3389/fimmu.2022.912876. PMID: 35874745.
Enlace PubMed
Resumen breve: Revisión centrada en mecanismos de daño tisular que no dependen de complemento, con especial atención a células efectoras y vías tipo 2.
-
Fang H, Zhang Y, Li N, et al. The Autoimmune Skin Disease Bullous Pemphigoid: The Role of Mast Cells in Autoantibody-Induced Tissue Injury. Front Immunol. 2018;9:407.
DOI: 10.3389/fimmu.2018.00407. PMID: 29545809.
Enlace PubMed
Resumen breve: Revisión sobre el papel de los mastocitos en PA, integrando datos experimentales de modelos animales y observaciones clínicas.
-
Lin L, Hwang BJ, Culton DA, et al. Eosinophils Mediate Tissue Injury in the Autoimmune Skin Disease Bullous Pemphigoid. J Invest Dermatol. 2018;138(5):1032-1043.
DOI: 10.1016/j.jid.2017.11.031. PMID: 29246800.
Enlace PubMed
Resumen breve: Estudio experimental que demuestra el papel central de los eosinófilos como mediadores de daño en PA, enlazando IgE, eosinofilia y formación de ampollas.
-
Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: A systematic review. J Am Acad Dermatol.2018;78(5):989-995.e2.
DOI: no disponible en resumen. PMID: 29102490.
Enlace PubMed
Resumen breve: Revisión sistemática sobre PA no ampolloso, describiendo fenotipos clínicos, retrasos diagnósticos y la necesidad de sospecha elevada en prurito crónico del anciano.
-
Lamberts A, Meijer JM, Diercks GFH, et al. Nonbullous pemphigoid: Insights in clinical and diagnostic findings, treatment responses, and prognosis. J Am Acad Dermatol. 2019;81(2).
DOI: no disponible en resumen. PMID: 31009674.
Enlace PubMed
Resumen breve: Estudio de cohorte que caracteriza en detalle la presentación clínica, respuesta al tratamiento y pronóstico de PA no ampolloso.
-
Meijer JM, Diercks GFH, de Lang EWG, et al. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA Dermatol. 2019;155(2):158-165.
DOI: 10.1001/jamadermatol.2018.4390. PMID: 30624575.
Enlace PubMed
Resumen breve: Estudio multicéntrico que define la estrategia diagnóstica óptima para PA (DIF, IIF salt-split, ELISA BP180) y establece requisitos mínimos diagnósticos.
-
Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134(9):1075-1080.
DOI: no disponible en resumen. PMID: 9762017.
Enlace PubMed
Resumen breve: Trabajo clásico que propone criterios clínicos (edad, ausencia de cicatrices atróficas, escasa afectación mucosa) que ayudan a predecir PA en ausencia inicial de pruebas inmunológicas avanzadas.
-
Kridin K, Bergman R, et al. Mortality in Patients with Bullous Pemphigoid: A Retrospective Cohort Study and Meta-Analysis. J Am Acad Dermatol. 2019;80(4):1168-1176.e1.
DOI: no disponible en resumen. PMID: 29963683.
Enlace PubMed
Resumen breve: Cohorte israelí y metaanálisis que cuantifican la mortalidad a 1 año en PA y analizan factores de mal pronóstico (edad, comorbilidades).
-
Liu SD, Chow S-KY, Chen YJ, et al. Association Between Medication Use and Bullous Pemphigoid: A Population-Based Case-Control Study. JAMA Dermatol. 2020;156(8):895-903.
DOI: no disponible en resumen. PMCID: PMC7301306.
Enlace PMC
Resumen breve: Estudio de base poblacional que analiza asociación de múltiples fármacos con PA, destacando gliptinas, diuréticos y fármacos neurolépticos como factores de riesgo significativos.
-
Feliciani C, Joly P, Jonkman MF, et al. Management of bullous pemphigoid: The European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol.2015;172(4):867-877.
DOI: 10.1111/bjd.13717. PMID: 25827742.
Enlace PubMed
Resumen breve: Primera guía de consenso europea que estandariza el manejo del PA, incluyendo recomendaciones sobre corticoides tópicos, sistémicos e inmunosupresores.
-
Borradori L, Van Beek N, Feliciani C, et al. Updated S2K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol.2022;36(10):1689-1704.
DOI: no disponible en resumen. PMID: 35766904.
Enlace PubMed
Resumen breve: Actualización de las guías europeas que refuerza el papel de los corticoides tópicos como primera línea y define algoritmos modernos de tratamiento escalonado.
-
Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346(5):321-327.
DOI: 10.1056/NEJMoa011592. PMID: 11821508.
Enlace PubMed
Resumen breve: Ensayo clínico aleatorizado que demuestra la superioridad en seguridad y eficacia de corticoides tópicos de alta potencia frente a prednisona oral en PA extenso.
-
Ruggiero A, Megna M, Camela E, et al. Strategies to Improve Outcomes of Bullous Pemphigoid: A Comprehensive Review of Clinical Presentations, Diagnosis, and Patients’ Assessment. Clin Cosmet Investig Dermatol. 2022;15:661-673.
DOI: 10.2147/CCID.S267573. PMID: 35444441.
Enlace PubMed
Resumen breve: Revisión que integra presentaciones clínicas, herramientas de evaluación de actividad (BPDAI, ABSIS) y recomendaciones para optimizar el seguimiento y la calidad de vida.
-
Granados-Betancort E, Sánchez-Díaz M, Muñoz-Barba D, Arias-Santiago S. Omalizumab and Dupilumab for the Treatment of Bullous Pemphigoid: A Systematic Review. J Clin Med. 2024;13(16):4844.
DOI: 10.3390/jcm13164844. PMID: 39200987.
Enlace PubMed
Resumen breve: Revisión sistemática de casos de PA tratados con omalizumab o dupilumab, mostrando altas tasas de respuesta y perfil de seguridad favorable en enfermedad refractaria.
-
Leisti P, Pankakoski A, Jokelainen J, et al. Accurate diagnosis of bullous pemphigoid requires multiple health care visits. Front Immunol. 2023;14:1281302.
DOI: 10.3389/fimmu.2023.1281302. PMID: 38090583.
Enlace PubMed
Resumen breve: Estudio que valida el código ICD-10 L12.0 y muestra que muchos pacientes requieren varias consultas para confirmar el diagnóstico de PA, destacando la importancia de múltiples contactos asistenciales.

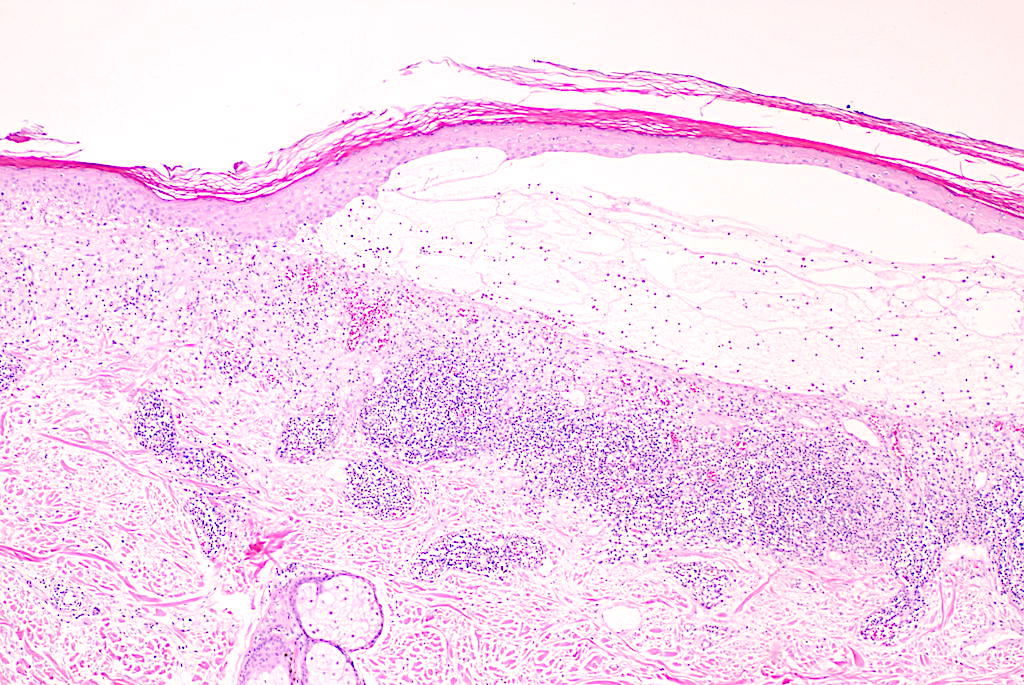
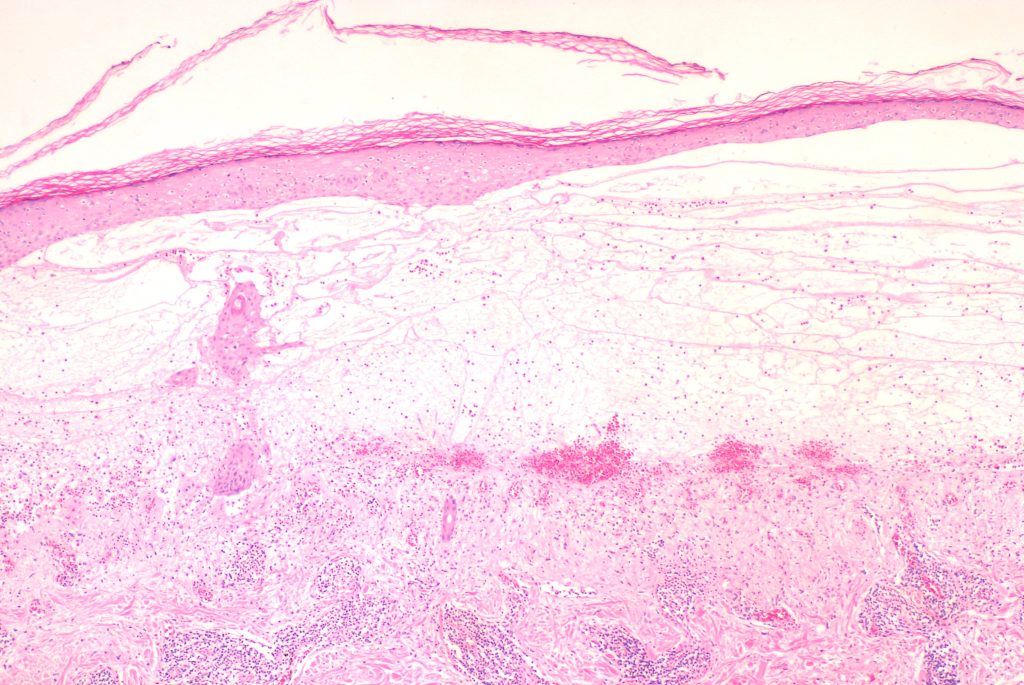
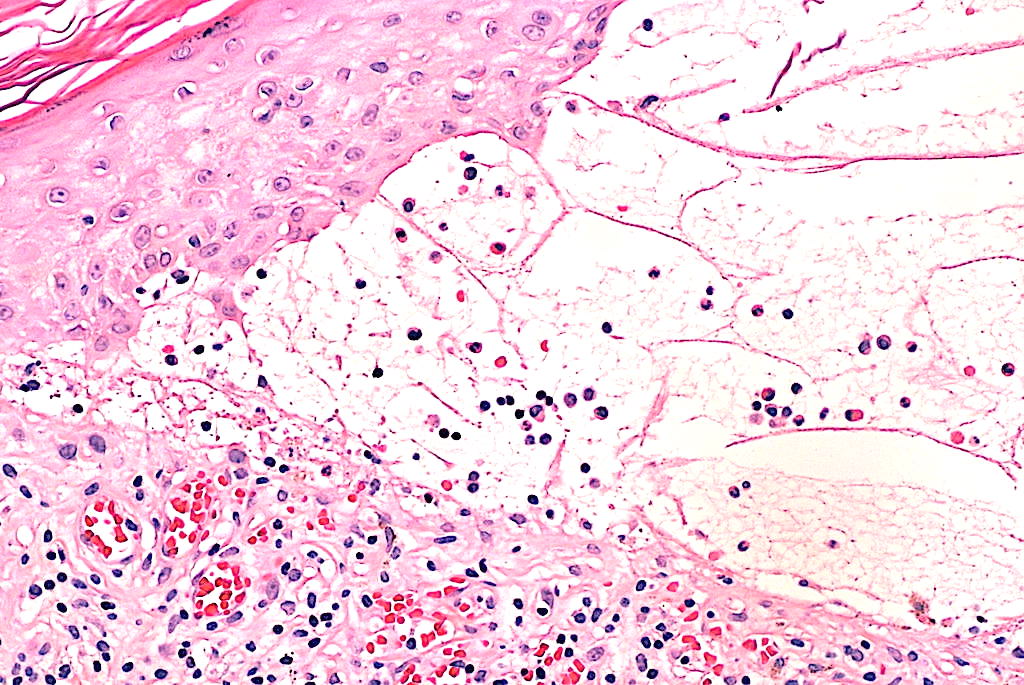
 |
|