
-
Nombres: Xantogranuloma juvenil (JXG); pertenece al grupo de las histiocitosis no Langerhans (NLCH).
-
Primera descripción: Adamson, 1905 (“congenital xanthoma multiplex”); la nomenclatura moderna se consolidó a mediados del siglo XX.
-
Epidemiología: Predomina en lactantes y niños pequeños; ≈ primera aparición en los dos primeros años de vida; puede ser congénito. Curso cutáneo típico benigno con involución espontánea.
-
Clasificación actual: Incluido en la familia de histiocitosis (grupo C/NLCH) y en tumores histiocíticos y dendríticos según la revisión de la Histiocyte Society/OMS.
-
Códigos: ICD-10: D76.3 (otras histiocitosis); ICD-11: 2B31.0 (Juvenile xanthogranuloma).
Buscar enfermedad
Creación:
27/11/2025
Última actualización:
09/12/2025
“Este contenido es exclusivamente informativo y está dirigido a profesionales de la salud. No reemplaza la valoración clínica ni el juicio médico, no establece relación médico-paciente y no constituye estándar de cuidado. La medicina evoluciona rápidamente: contraste siempre las fuentes y utilice esta información solo como un insumo adicional para la toma de decisiones.”
General
- Proliferación de histiocitos (dermales/factor XIIIa+) con respuesta inflamatoria variable; en la mayoría de los casos cutáneos es autolimitado. Alteraciones activadoras en vías MAPK/ERK y, en subgrupos extracutáneos, alteraciones CSF1R, ALK y fusiones CLTC::SYK/NTRK1 han sido descritas (implicación terapéutica en casos sistémicos/atípicos).
-
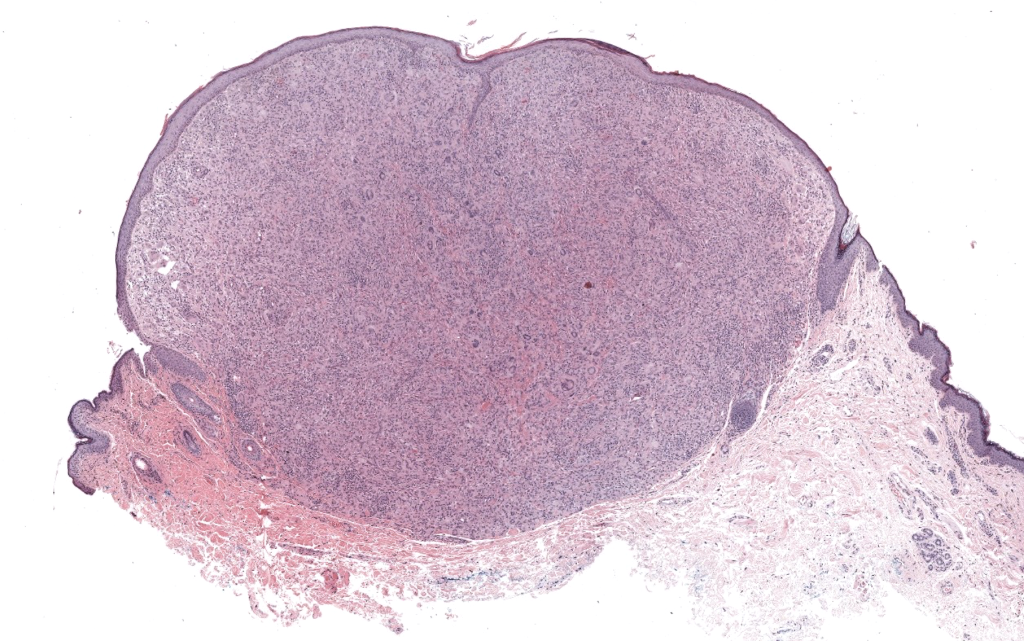
Lesión elemental: Pápula o nódulo cúpuliforme, firme, bien delimitado, color amarillo-anaranjado/eritematoso; a veces telangiectasias superficiales. “Eruptivo” cuando son múltiples.
-
Zonas típicas: Cara, cuello, tronco; puede afectar mucosa (oral), subcutis, e, infrecuentemente, vísceras/CNS (sistémico).
-
Clínica prototípica: Niño <2 años con lesión única; evolución de rojiza a amarilla, con involución en 1–5 años dejando hiperpigmentación o leve atrofia.
-
Hallazgos complementarios: Dermoscopia con patrón “sol poniente” (disco central amarillo-anaranjado con halo eritematoso) y vasos lineales/ramificados; no es exclusivo de JXG.
-
Evolución: Autolimitado en la mayoría de formas cutáneas; las formas profundas/múltiples pueden persistir; el JXG ocular (iris) puede producir hipema/glaucoma.
-
Formas atípicas: Gigante/placa, múltiple/diseminado, subcutáneo/intramuscular (“profundo”), mucoso (oral), visceral, SNC-JXG (Sistema Nervioso Central)
- Nevus de Spitz/ Nevus Melanocítico Spitzoide,
- mastocitoma,
- dermatofibroma,
- xantomas (dislipidemias),
- hemangioma,
- neurofibroma,
- histiocitosis de células de Langerhans (LCH).
-
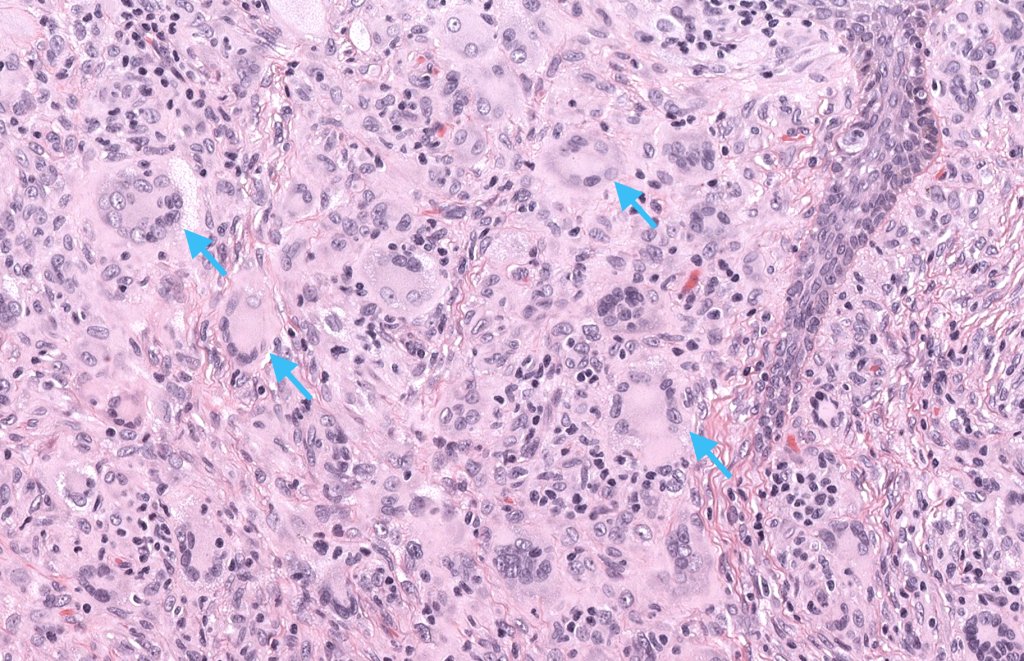
Patrón: Dermatitis granulomatosa/xantogranulomatosa con histiocitos espumosos y células gigantes de Touton; infiltrado linfoeosinofílico variable; epidermis habitualmente respetada.
-
IHQ (inmunohistoquímica): CD68, CD163, factor XIIIa, fascina positivos / CD1a, langerina (CD207) negativos / S100 usualmente negativo o débil (puede ser positivo en raros casos).
-
Tinciones especiales/DIF (inmunofluorescencia directa): No necesarias de rutina.
-
Pruebas moleculares (cuando atípico/sistémico): Búsqueda de BRAFV600E (más relevante en CNS-JXG/entidades solapadas), ALK (FISH/NGS), CSF1R, NTRK1, fusiones CLTC::SYK.
- Histiocitosis de Células de Langerhans (LCH) (CD1a+/langerina+, Birbeck),
- Reticulohistiocitoma
- Erdheim-Chester, Rosai-Dorfman (S100++ con emperipolesis),
- Dermatofibroma (factor XIIIa+, pero sin Touton ni xantomización típica),
- Xantogranuloma necrobiótico
Cutáneo solitario típico: No requiere estudios adicionales.
Múltiples/atípicos/síntomas sistémicos: Biometría hemática, PFH, US abdominal, Rx tórax; RM dirigida si síntomas neurológicos; oftalmología si <2 años con lesiones múltiples o en cabeza/cuello o cualquier signo ocular. El cribado universal ocular en lesiones únicas y alejadas del ojo no es necesario.
Biopsia:
-
Tipo: Punch profundo o escisional incluyendo dermis y subcutis superficial; ideal en lesión “activa” (no involutiva).
-
Consideraciones: Suspender tópicos irritantes previos si es posible; evitar zonas ulceradas/antiguas; fijación en formalina 10% para HE/IHQ; no precisa IFD de rutina.
-
Contraindicaciones: Relativas (coagulopatía, localización ocular con riesgo de hipema).
Primera línea (cutáneo típico): Observación expectante; educación familiar. Exéresis si diagnóstico incierto, sangrado, ulceración o impacto estético/funcional.
Opciones locales (seleccionados): Láser (p. ej., CO₂/ablativo o colorante pulsado) en variantes especiales (placa gigante, secuelas) y exéresis; evidencia basada en series/casos.
-
JXG ocular: Corticoides tópicos/perioculares/sistémicos, manejo del hipema/glaucoma; cirugía si refractario. Pronóstico visual mejor con tratamiento precoz.
-
JXG sistémico/extracutáneo (SJXG): Esquemas tomados de LCH (p. ej., vinblastina + prednisona), citarabina, cladrribina; sirolimus oral/ tópico (datos de casos/series); terapias dirigidas si mutaciones diana (p. ej., inhibidores ALK como alectinib en ALK-+; imatinib reportado en CSF1R mutado CNS-JXG). Individualizar en centros con experiencia.
-
Cuidados/seguridad: Vinblastina (neuro/mielotoxicidad), citarabina (mielosupresión), cladribina (linfopenia/infecciones), sirolimus (dislipidemia, estomatitis, inmunosupresión), ALK-i (hepatotoxicidad, bradicardia), imatinib (edema, hepatotoxicidad, citopenias). (Basado en fichas y series; seleccionar según mutaciones y extensión).
-
Signos oculares (fotofobia, epífora, hipema): derivar a oftalmología de inmediato.
-
Lesiones múltiples, profundas o síntomas sistémicos (fiebre, pérdida ponderal, hepatoesplenomegalia, síntomas neurológicos): solicitar estudios básicos y de imagen.
-
Niño con NF1 (neurofibromatosis tipo 1) + JXG: asociación descrita con JMML (leucemia mielomonocítica juvenil); aunque el riesgo absoluto es bajo y debatido, mantener alta vigilancia clínica y hematológica.
-
JXG en SNC o visceral: manejo en unidades con experiencia en histiocitosis pediátrica/hemato-onco; considerar perfil molecular (posible terapia dirigida).
-
Touton + CD68/CD163/factor XIIIa positivos y CD1a/langerina negativos orientan fuertemente a JXG vs LCH.
-
El patrón dermatoscópico “sol poniente” apoya el diagnóstico pero no es exclusivo.
-
La mayoría de lesiones cutáneas involucionan sin tratamiento en 1–5 años.
-
Dehner LP. Juvenile xanthogranulomas in the first two decades of life… Am J Surg Pathol. 2003;27:579–593. DOI: no disponible · **PMID:**12717244. Resumen: Serie de 174 casos que caracteriza espectro cutáneo y extracutáneo, morfología (Touton), curso y complicaciones.
-
Hernández-Martín A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol.1997;36:355–367. **DOI:**10.1016/S0190-9622(97)80207-1 · **PMID:**9091465. Resumen: Revisión clínica clásica de presentación, evolución e histología del JXG.
-
So JTY, et al. Juvenile xanthogranuloma: a review with updates on the pathogenesis, associations… Pediatr Dermatol.2020;37:xxx–xxx. DOI: no disponible · **PMID:**32468628. Resumen: Revisión moderna con recomendaciones de cribado/seguimiento y manejo.
-
Emile JF, et al. Revised classification of histiocytoses… Blood. 2016;127:2672–2681. **DOI:**10.1182/blood-2016-01-690636 · **PMID:**26966089. Resumen: Marco clasificatorio actual de histiocitosis (incluye JXG).
-
Tahan SR, et al. Juvenile xanthogranuloma. J Am Acad Dermatol. 1989;20:xxx–xxx. DOI: no disponible · **PMID:**2505733. Resumen: Caracterización clínico-patológica de lesiones cutáneas típicas.
-
Nascimento AF, et al. Juvenile xanthogranuloma: comparative clinicopathologic study… Am J Surg Pathol.1999/1997. DOI: no disponible · **PMID:**9199641. Resumen: Subtipos profundos (subcutáneo/intramuscular) y su histología diferencial.
-
Newman CC, et al. Nonlipidized juvenile xanthogranuloma… Am J Surg Pathol. 1997;21:xxx–xxx. DOI: no disponible · **PMID:**9144693. Resumen: Variante no lipidizada que puede simular neoplasias agresivas; IHQ clave.
-
Samara WA, et al. Juvenile Xanthogranuloma of the iris: clinical features/outcomes… Ophthalmology. 2015;122:xxxx. DOI: no disponible · **PMID:**26189188. Resumen: Serie oftalmológica amplia: complicaciones (hipema, glaucoma) y resultados visuales.
-
Freyer DR, et al. Juvenile xanthogranuloma: forms of systemic disease… J Pediatr. 1996;129:227–237. DOI: no disponible · **PMID:**8765620. Resumen: Descripción clásica del SJXG, distribución orgánica y consideraciones terapéuticas.
-
Zou T, et al. Systemic juvenile xanthogranuloma: a systematic review. Pediatr Blood Cancer. 2023;70:e30232. **DOI:**10.1002/pbc.30232 · **PMID:**36779547. Resumen: Revisión sistemática moderna de SJXG: perfiles, tratamientos y resultados.
-
Kemps PG, et al. Recurrent CLTC::SYK fusions and CSF1R mutations in JXG of soft tissue. Blood. 2024;144:2439–2455. **DOI:**10.1182/blood.2024025127 · **PMID:**39316650. Resumen: Cohorte con alteraciones diana frecuentes en JXG extracutáneo; implicaciones terapéuticas.
-
Durham BH, et al. Activating mutations in CSF1R… Nat Med. 2019;25:1839–1842. **DOI:**10.1038/s41591-019-0653-6 · **PMID:**31768065. Resumen: Mutaciones en CSF1R/RTKs en histiocitosis; respuestas a inhibidores selectivos.
-
Picarsic J, et al. BRAFV600E in CNS-JXG: revised algorithm… Acta Neuropathol Commun. 2019;7:168. **DOI:**10.1186/s40478-019-0811-6 · **PMID:**31685033. Resumen: Alta tasa de BRAF en JXG del SNC pediátrico; guía diagnóstica.
-
Xu J, et al. Systemic JXG has higher frequency of ALK translocations than BRAFV600E. J Am Acad Dermatol.2023;88:656–659. **DOI:**10.1016/j.jaad.2020.08.053 · **PMID:**32822792. Resumen: Señala papel de ALK en SJXG y opción de terapias ALK-i.
-
Pretel M, et al. Dermoscopic “setting sun” pattern of JXG. J Am Acad Dermatol. 2015;72:S73–S75. **DOI:**10.1016/j.jaad.2014.09.042 · **PMID:**25500052. Resumen: Describe el patrón dermatoscópico clásico.
-
Xu J, et al. Dermoscopic patterns in JXG (serie n=41). Front Med (Lausanne). 2021;7:618946. **DOI:**10.3389/fmed.2020.618946 · **PMID:**33521026. Resumen: Correlación dermatoscopia-histología por estadio; utilidad en seguimiento.
-
Toker M, et al. Oral sirolimus for JXG (2 casos). [Revista] 2024. DOI: no disponible · **PMID:**38444069. Resumen: Respuesta clínica rápida; sugiere papel del eje mTOR en JXG.
-
Martínez-Torres V, et al. CNS-JXG con CSF1R mutado, respuesta a imatinib. Neuro Oncol. 2024. DOI: no disponible · PMID: no disponible (PMCID disponible). Resumen: Primera respuesta sostenida a imatinib en CSF1R-mutado del SNC.
-
Samara WA, et al. (reiterado en 8) – manejo y outcomes oculares. (ver 8)
-
Höck M, et al. Various clinical spectra of JXG: 2 casos + revisión. BMC Pediatr. 2019;19:xxx. **DOI:**10.1186/s12887-019-1490-y · PMID: no disponible en página, indexado en PubMed Central. Resumen:Amplía espectro, inmunofenotipo e imagen.
 |
|